
ETAP DRUGI – analiza składu chemicznego osadu
Metodyka badań
W celu poznania składu chemicznego badanego materiału przeprowadzono następujące analizy:
Oznaczanie suchej substancji przy użyciu wagosuszarki [PN-78/A-74701]
Oznaczanie zawartości popiołu ogólnego [ICC STANDARD No.104/1]
Oznaczanie kwasowości [PN-78/A-74701]
Oznaczanie pH [PN-78/A-74701]
Oznaczanie zawartości chlorków metodą Mohra [PN-78/A-74701]
Oznaczanie zawartości tłuszczu metoda Soxhleta [PN- EN ISO 3947]
W odtłuszczonej zlewce sporządzono zawiesinę około 10 g (z dokładnością do ±0,0001 g) badanej próbki w 100 cm3 wody destylowanej. Osobno sporządzono roztwór 100 cm3 stężonego kwasu solnego w 200 cm3 wody destylowanej. Roztwór ten ogrzano do wrzenia i wlano do zawiesiny. Mieszaninę ogrzewano przez 45 minut, aż do zaniku reakcji jodowo skrobiowej i sączono przez sączek karbowany, który następnie suszono w suszarce w temperaturze 105 ºC w ciągu 4 godzin. Wysuszony sączek zawierający tłuszcz przeniesiono do nasadki ekstrakcyjnej aparatu Soxhleta i prowadzono ekstrakcję przy użyciu eteru naftowego. Po zakończeniu ekstrakcji oddestylowano rozpuszczalnik, natomiast kolbę wysuszono w suszarce w temperaturze 105 ºC w czasie 60 minut, po czym schłodzono ją w eksykatorze i zważono.
Procentową zawartość surowego tłuszczu (X) w suchej substancji obliczono wg wzoru:
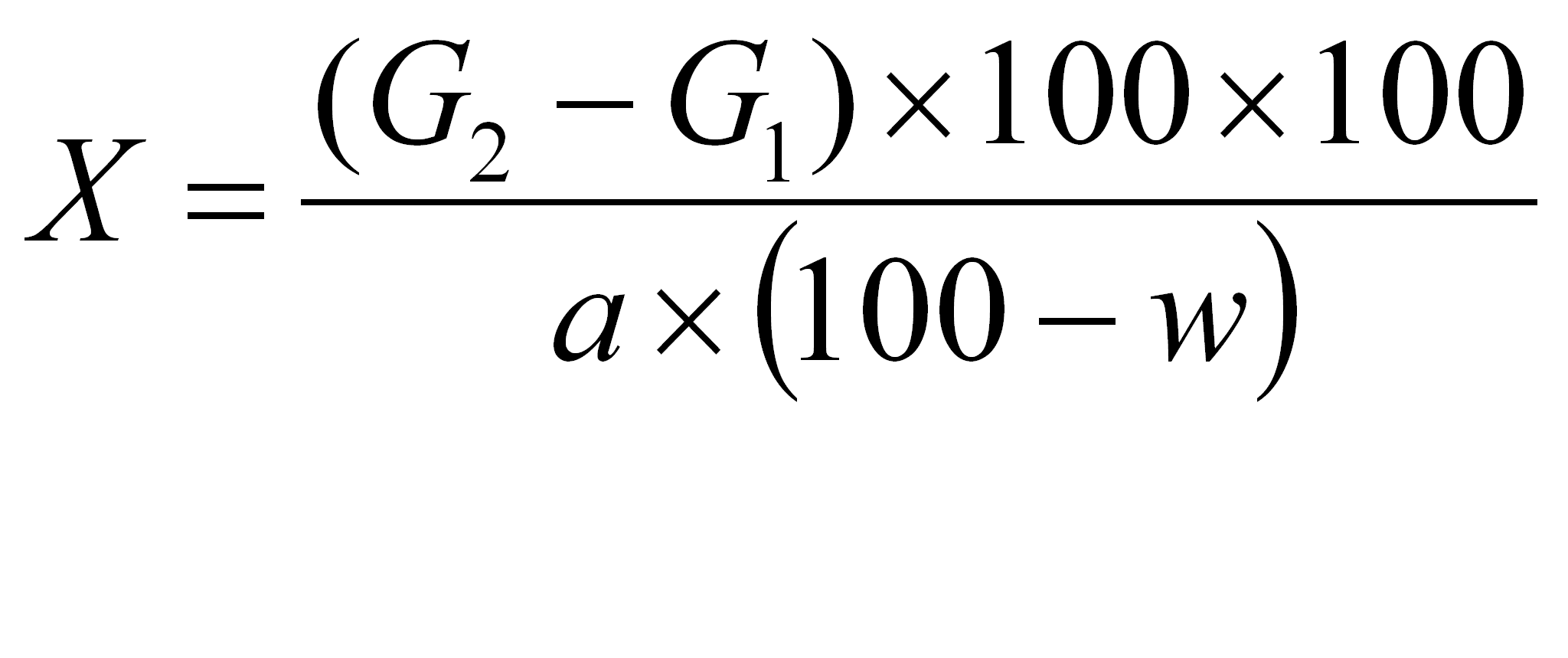
gdzie:
G1 - masa pustej kolby (g),
G2 - masa kolby z tłuszczem (g),
w - zawartość wody w próbce (%),
a - naważka badanej substancji (g).
Oznaczanie zawartości białka metodą Kjeldahla [ICC STANDARD No.105/2]
Odważono z dokładnością do ±0,0001 g próbkę o masie ok. 2 g i przeniesiono ilościowo do kolby Kjeldahla, dodano 15 cm3 stężonego kwasu siarkowego na 1 g suchej próbki i 0,5 g mieszaniny selenowej. Kolbę z badanym materiałem poddano mineralizacji przy zakresie temperatury 360 – 380 ºC. Spalanie prowadzono do chwili uzyskania jasnozielonego, klarownego zabarwienia roztworu. Po zakończeniu mineralizacji próbkę przeniesiono do kolby destylacyjnej i zobojętniono 33 % roztworem ługu sodowego w obecności fenoloftaleiny jako wskaźnika. Następnie przeprowadzono destylację z parą wodną. Destylat zbierano w odbieralniku, w którym znajdowało się 25 cm3 0,1 M kwasu solnego i kilka kropli wskaźnika Tashiro. Destylację prowadzono ok. 20 minut do uzyskania dwukrotnej ilości destylatu. Otrzymany destylat miareczkowano 0,1 M roztworem wodorotlenku sodowego do wystąpienia barwy seledynowej. Równolegle wykonano próbę ślepą.
Procentową zawartość białka (X) w suchej substancji obliczono wg wzoru:
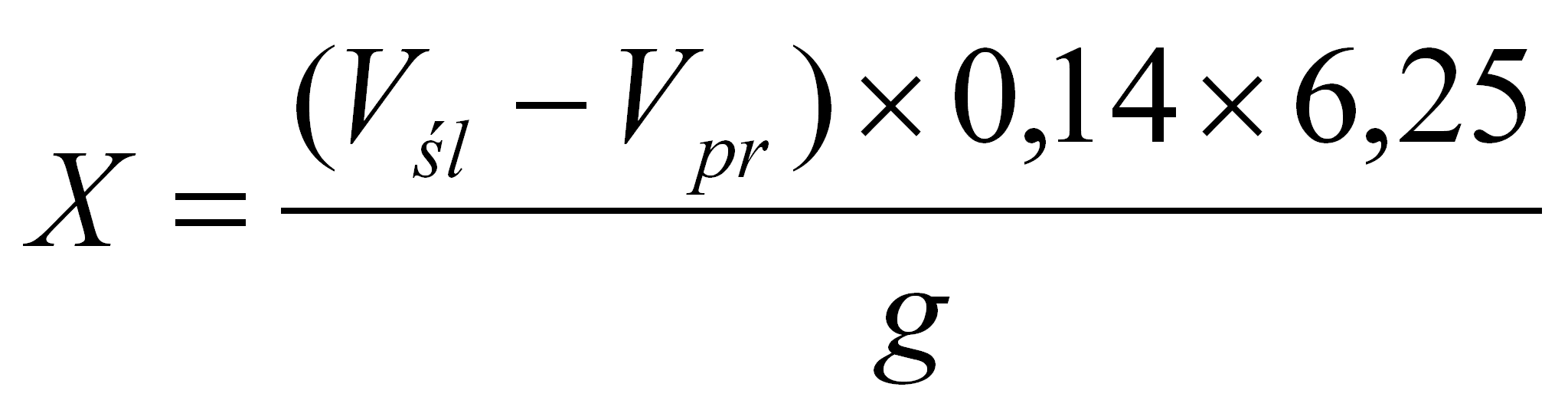
gdzie:
Vśl - objętość 0,1 M roztworu wodorotlenku sodowego zużytego do miareczkowania próby ślepej (cm3),
Vpr - objętość 0,1 M roztworu wodorotlenku sodowego zużytego do miareczkowania próby właściwej (cm3),
g - naważka badanej próbki w przeliczeniu na suchą masę (g),
0,14 - ilość azotu (mg), której odpowiada 1 cm3 0,1 M kwasu solnego,
6,25 - mnożnik białkowy dla zboża.
Oznaczanie zawartości β-glukanów [AOAC 995.16]
Próbkę hydrolizatu w ilości 8 – 12 g z dokładnością do ±0,0001 g odważono i wsypano do szklanej probówki wirówkowej. Nawilżono 0,2 cm3 wodnego roztworu etanolu (50 %) dla ułatwienia rozpuszczenia. Dodano 4 cm3 buforu fosforanowo sodowego i wymieszano w wytrząsarce Vorteks firmy Kartell. Po wymieszaniu próbkę umieszczono we wrzącej łaźni wodnej i termostatowano przez 1 minutę, następnie energicznie wytrząsano w wytrząsarce, termostatowano przez 2 minuty w temperaturze 100 ºC i ponownie wytrząsano. Po procesie wytrząsania próbkę inkubowano w 50 ºC dla wyrównania temperatury przez okres 5 minut.
Po dodaniu 0,2 cm3 lichenazy próbkę inkubowano przez 60 minut w temperaturze 50 ºC, od czasu do czasu wytrząsając. Dodano następnie 5 cm3 buforu sodowego i energicznie wymieszano w wytrząsarce. Po czasie 5 min, kiedy to próbkę pozostawiono w temperaturze pokojowej, wirowano ją przy 1000 obr/min przez 10 minut.
Do trzech probówek ostrożnie przeniesiono 0,1 cm3 otrzymanego roztworu przy użyciu dozownika Rainin EDP-2, do dwóch probówek dodano 0,1 cm3 β-glukozydazy rozpuszczonej w 0,05 M buforu octanu sodu (pH 4.0), a do trzeciej (próba ślepa) 0,1 cm3 0,05 M buforu octanowego (pH 4.0). Wszystkie probówki inkubowano przez 10 minut w temperaturze 50 ºC.
Do każdej probówki dodano 3,0 cm3 reagenta (glukozo/oksydazo/peroksydazy) firmy Megazyme i inkubowano przez 20 minut w temperaturze 50 ºC. Po wyjęciu próbek z łaźni wodnej napełniono badanym roztworem kuwetę i zmierzono absorpcję przy długości fali λ= 510 nm.
Procentową zawartość β-glukanów przeliczono na suchą substancję.
Oznaczanie rozpuszczalnej i nierozpuszczalnej ilości błonnika pokarmowego [AOAC 991.43]
Przygotowanie materiału do oznaczenia
Próbkę osadu po hydrolizie naważono w dwóch powtórzeniach po 1,000 g ± 0,005 g w zlewkach na 400 cm3. Dodano po 40 cm3 buforu fosforanowego (pH 6) i mieszano do równomiernego rozprowadzenia próbki. Przeprowadzono inkubacje z α-amylazą. Do próbek dodano po 5 cm3 termostabilnej α-amylazy, wymieszano, zlewki przykryto folią aluminiową, i wytrząsano w łaźni wodnej o temperaturze 95-100 ºC przez 35 min. Po tym czasie ochłodzono próbkę do temperatury 60 ºC, zebrano osad ze ścianek zlewki i przepłukano szpatułkę 10 cm3 wody. Następnie dodano po 100 cm3 roztworu proteazy, przykryto zlewki folią aluminiową i wytrząsano w łaźni wodnej w temperaturze 60 ºC przez 30 min (czas mierzono od osiągnięcia w zlewce temperatury 60 ºC). Próbki wyjęto z łaźni, dodano po 5 cm3 0,561 M HCl i ustalono pH w zakresie 4,1 - 4,8 przy użyciu 5 % roztworu NaOH lub 5 % HCl. Po ustaleniu odpowiedniego pH przeprowadzono inkubacje z amyloglukozydazą. Dodano po 200 cm3 enzymu ciągle mieszając, przykryto folią aluminiową i inkubowano w łaźni wodnej w temperaturze 60 ºC przez 60 min.
Nierozpuszczalna frakcja błonnika (IDF)
Zważono tygle z celitem z dokładnością do 0,0001 g, złoże celitu zwilżono 3 cm3 wody i odsączono pod próżnią do wyrównania powierzchni. Badany roztwór powstały po inkubacji z enzymami przesączono, a następnie przemyto 2 krotnie 10 cm3 wody destylowanej o temperaturze 70 ºC. Przesącz zachowano w wytarowanej zlewce na 600 cm3 do oznaczenia frakcji rozpuszczalnej błonnika. Osad w tyglu przemywano 2 krotnie 10 cm3 95 % etanolu i 10 cm3 acetonu. Tygle z osadem suszono w temperaturze 103 ºC przez całą noc. Po wysuszeniu i ostudzeniu, tygle zważono z dokładnością do ± 0,0001 g. Obliczono masę błonnika odejmując masę tygla z celitem. Oznaczono zawartość białka w osadzie z pierwszej próbki (metodą Kjedahla) oraz zawartość popiołu w osadzie z drugiej próbki (temperatura 525 ºC przez 5 godzin). Po odjęciu od całości błonnika oznaczonej zawartości białka i popiołu otrzymano rzeczywistą zawartość błonnika nierozpuszczalnego.
Rozpuszczalna frakcja błonnika (SDF)
Przesącz w zlewce, który powstał po przemywaniu osadu wykorzystano do oznaczania nierozpuszczalnej frakcji błonnika. Przesącz po filtracji zważono. Dodano 4 objętości 95 % etanolu ogrzanego do temperatury 60 ºC, popłukując kolbę filtracyjną alkoholem. Zostawiono do wytrącenia osadu na 1 godzinę. Zważono tygle z celitem z dokładnością do ± 0,0001 g, zwilżono warstwę celitu 15 cm3 78 % etanolu i odsączono pod próżnią dla wyrównania powierzchni. Roztwór przesączono przenosząc ilościowo osad przy użyciu 78 % etanolu i gumowej szpatułki. Osad przemyto 2 krotnie 15 cm3 78 % i 95 % etanolu oraz 2 krotnie 15 cm3 acetonu. Tygle z osadem suszono w temperaturze 103 ºC przez całą noc. Po wysuszeniu tygle studzono w eksykatorze przez 1 godzinę, a następnie zważono z dokładnością do ± 0,0001 g. Obliczono masę błonnika odejmując masę tygla z celitem. Oznaczono zawartość białka w osadzie z pierwszej próbki (metodą Kjedahla) oraz zawartość popiołu w osadzie z drugiej próbki (temperatura 525 ºC przez 5 gdzin). Po odjęciu od całości błonnika oznaczonej zawartości białka i popiołu otrzymano rzeczywistą zawartość błonnika rozpuszczalnego.
Oznaczanie zawartości skrobi metodą polarymetryczną wg Clendenninga
[ICC STANDARD No.122]
Z badanego materiału odważono 2-5 g do probówki wirówkowej na 100 cm3, dodano 20 cm3 alkoholowego roztworu chlorku rtęciowego. Wymieszano dokładnie pręcikiem szklanym (homogenizacja 2 min.), po czym pręcik spłukano 20 cm3 roztworu chlorku rtęciowego i wirowano przez 20 min. przy prędkości 4-6 tys. obr/min. Klarowny roztwór znad osadu zdekantowano (roztwór odrzucono), dodano 5 cm3 wody destylowanej, wymieszano a następnie pręcik przemyto 5 cm3 wody destylowanej. Osad z probówki wirówkowej przeniesiono ilościowo do kolby stożkowej na 250 cm3 przy pomocy 60 cm3 chlorku wapnia o gęstości 1,3 g/cm3, dodano kilka kuleczek szklanych. Następnie ostrożnie gotowano przez 30 minut pod chłodnicą zwrotną, po upływie tego czasu kolbkę ostudzono w strumieniu zimnej wody.
Zawartość kolbki (bez kuleczek szklanych) przeniesiono ilościowo do kolbki miarowej na 100 cm3. Kolbkę stożkową przemyto 4 razy po 5 chlorkiem wapnia o gęstości 1,3 g/cm3, a następnie zawartość kolbki miarowej odbiałczano przez dodanie 5 cm3 roztworu Carreza I i po dokładnym wymieszaniu 5 cm3 roztworu Carreza II. Zawartość kolby uzupełniono do kreski roztworem chlorku wapnia i po wymieszaniu odstawiono na 10 min. Następnie całość przesączono przez sączek karbowany odrzucając pierwsze partie przesączu. Rurkę polarymetryczną napełniono badanym roztworem, umieszczono w Polamacie i dokonano pomiaru przy długości fali λ= 546 nm.
Po dokonaniu wszystkich odczytów zawartość skrobi (ZS) w badanej próbce w przeliczeniu na suchą masę obliczono ze wzoru:
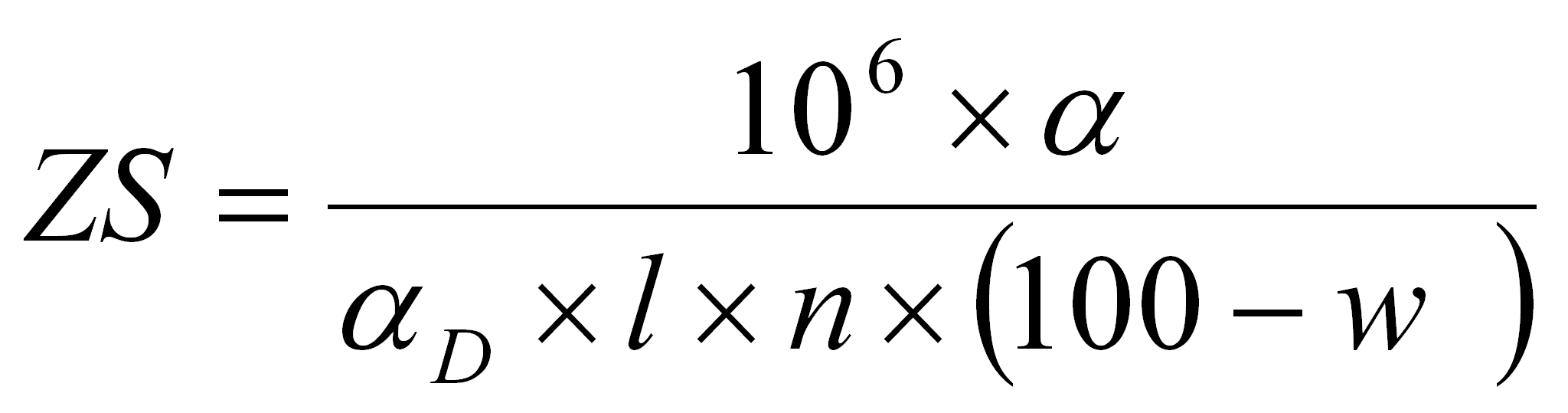
gdzie:
α - skręcalność roztworu w stopniach kątowych
αD - skręcalność właściwa = +240 º dla wszystkich skrobi
l - długość rurki polarymetrycznej =2 dm [dm]
n - naważka [g]
w - zawartość wody [%]
Oznaczanie zawartości cukrów redukujących metodą Luffa-Schoorla
[ICC STANDARD EEC/79/796]
Odważono 2 g próbki z dokładnością do ±0,0001 g, próbkę przeniesiono do kolby miarowej o pojemności 100 cm3, dodano około 50 cm3 gorącej wody i utrzymywano wysoką temperaturę ciągle mieszając, do uzyskania jednolitej mieszaniny. Następnie dodano 5 cm3 odczynnika Carreza I i 5 cm3 odczynnika Carreza II, wymieszano, uzupełniono kolbę wodą destylowaną do kreski i sączono przez karbowany sączek. Pobrano 25 cm3 przesączu, dodano 25 cm3 odczynnika Luffa- Schoorla. Gotowano pod chłodnicą zwrotną do wrzenia i utrzymywano tą temperaturę przez 10 minut. Następnie ochłodzono próbkę w strumieniu zimnej wody, dodano 10 cm3 jodku potasu i 25 cm3 3M kwasu siarkowego (IV). Miareczkowano 0,1 M tiosiarczanem sodu do odbarwienia wobec skrobi jako wskaźnika.
Równocześnie wykonano próbę ślepą używając 25 cm3 wody zamiast badanej próbki. Zawartość cukrów redukujących w badanej próbce obliczono z różnicy objętości 0,1 M tiosiarczanu sodu zużytego do miareczkowania próby badanej i próby ślepej. Na tej podstawie z odpowiednich tablic odczytano zawartość glukozy w badanej próbce i przeliczono ją na 100 g próbki.
komentarze
Copyright © 2008-2010 EPrace oraz autorzy prac.